Our Work
In eukaryotic cells, biochemical processes occur in specialized membrane-enclosed compartments - organelles - whose integrity must be preserved to ensure cellular health. Recently, it was discovered that mitochondria - the cellular power plants – establish physical contacts with a myriad of other organelles, with crucial functional implications. In particular, mitochondria are tethered to the endoplasmic reticulum, peroxisomes, lysosomes, to the plasma membrane and to themselves.
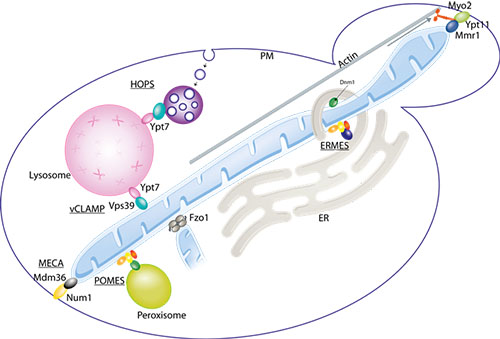
In eukaryotes, physical contacts between mitochondria and other organelles allows optimized performance. Mitochondria associate with each other (allowing their fusion), with the endoplasmic reticulum (ER - the production site for lipids and membrane proteins), the peroxisomes (necessary for several catabolic and anabolic reactions e.g. lipid synthesis and detoxification of reactive oxygen species), the lysosomes (where waste materials within the cell are digested to reusable building blocks) and the plasma membrane (PM - which separates each cell from its outside environment). The respective tethers are called MECA, ERMES, POMES and vCLAMP.
Mitochondria are very dynamic organelles that constantly fuse and divide in order to maintain a healthy network within the cell.
In our group we study mitochondrial fusion. This process is carried out by mitofusins, which are dynamin-related large GTPases located at the outer membrane of mitochondria, facing the cytoplasm. Mammals have two homologous mitofusins, Mfn1 and Mfn2, and the yeast mitofusin is called Fzo1.
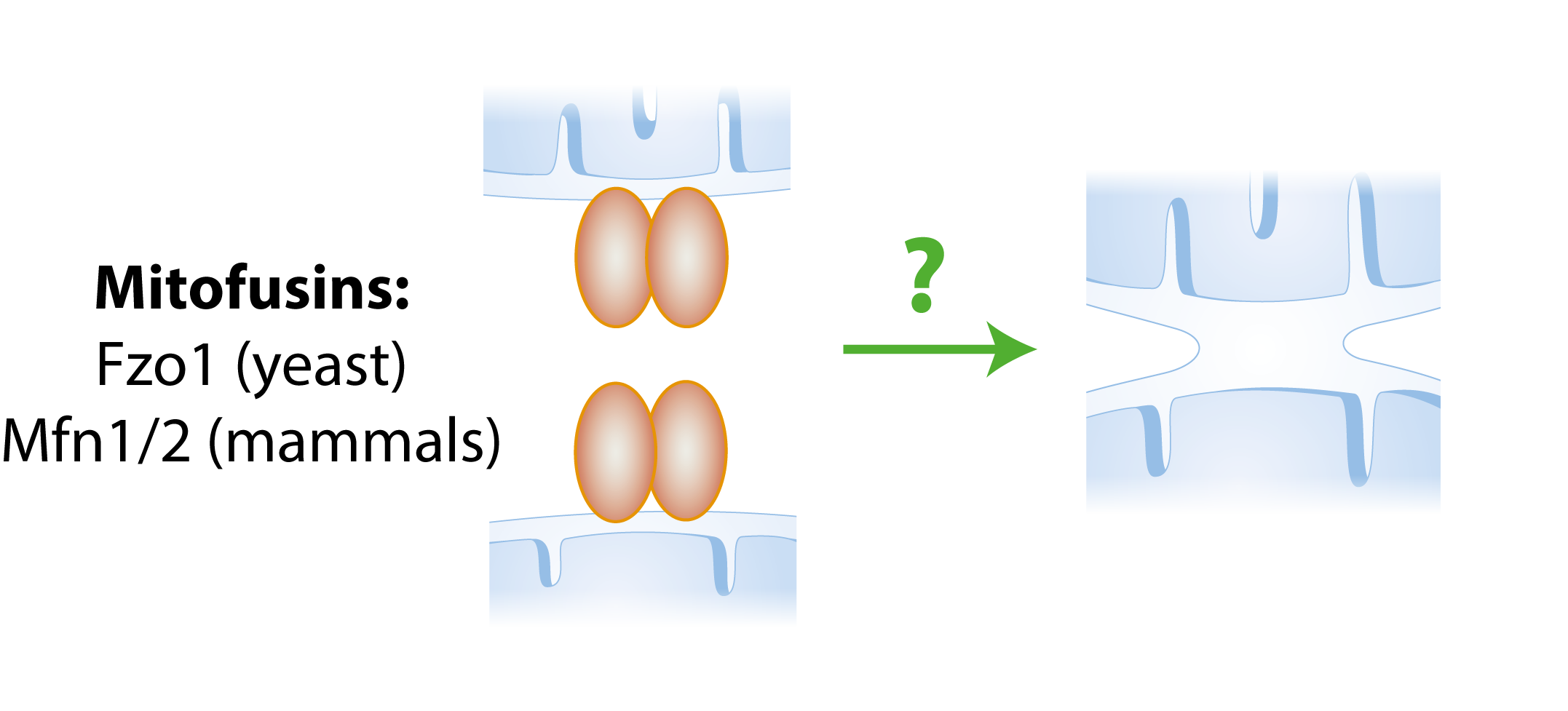
Simple fusion model. The presence of mitofusins on both mitochondria allows two mitochondria to fuse with each other.
Deficiencies in Mfn2 cause Charcot–Marie–Tooth disease (CMT), the most common degenerative disorder of the peripheral nervous system, and were also linked to Parkinson's disease (PD), the most frequent movement disorder. CMT is characterized by progressive distal weakness and muscular atrophy and sensory abnormalities. The subtype Charcot–Marie–Tooth type 2A (CMT2A) is caused by mutations in the mitochondrial gene Mfn2. Given that no effective therapy is available, it is particularly important to understand at the molecular basis how Mfn2 mutations contribute to the pathophysiologic mechanism of the CMT2A disease.

Bramwell 1907
Charcot–Marie–Tooth. The progressive distal weakness and muscular atrophy with feet exhibiting a high arch and claw toes are signs of this genetic disease.
