Mitochondrial Dynamics
In most cell types and organisms, mitochondria display a highly dynamic tubular network, constantly remodelled by opposing fusion and fission events. This plasticity is important to retain organelle function and integrity. Many neurodegenerative diseases demonstrate abnormal mitochondrial morphology and biochemical dysfunction.
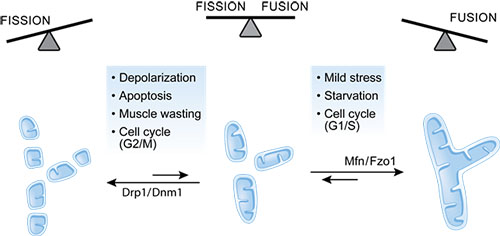
Mitochondria are very dynamic organelles that constantly fuse and divide. This plasticity is crucial for embryonic development, for neuronal survival, and is associated with mitophagy of damaged mitochondria and apoptosis (EMBO Rep 2014 15(3) 231-243).
Fusion facilitates mitochondrial content mixing and protects the cell against mitochondrial DNA loss, assuring maximal ATP production. Fission allows the proper distribution of mitochondria throughout the cell and also contributes to the separation of damaged mitochondria from the network, allowing their selective degradation. Block of fusion or fission severely impacts on mitochondrial morphology. A loss of fusion leads to fragmented mitochondria caused by unopposed fission, whereas fission deficiency results in a highly interconnected network due to ongoing fusion.
Impairment of mitochondrial morphology and function has been linked to several neurodegenerative diseases.
Fusion and fission of mitochondria and also of other cellular compartments depends on dynamin-related proteins (DRPs). DRPs are a special class of GTPases, which by self-association provide the mechanical forces necessary for membrane remodelling. Thereby, DRPs differ from conventional GTPases, which act as molecular switches by cycling between an active GTP-bound and an inactive GDP-bound state.
The fusion of mitochondria is mediated by conserved DRPs. OM fusion relies on the activity of mitofusins, whereas IM fusion requires Opa1/Mgm1. Mitochondrial fission depends on the dynamins Drp1 and Dyn2. Mutations in the mitofusin Mfn2 cause Charcot-Marie-Tooth type 2A neuropathy (CMT2A) and mutations in Opa1 cause autosomal dominant optic atrophy (ADOA). However, the molecular mechanisms leading to neurodegeneration are still largely unclear.